
La Sindrome di Sotos, chiamata anche Gigantismo cerebrale, Sindrome di Sotos-Dodge o Sequenza di Sotos, la malattia è stata descritta per la prima volta nel 1964 dal Dottor Sotos. È caratterizzata da una crescita al di sopra del normale e da ritardo nell’acquisizione delle principali tappe dello sviluppo. La Sindrome di Sotos non è però incompatibile con la vita e chi ne è affetto vive in modo pressoché normale.
Cos’è la sindrome di Sotos e come si manifesta
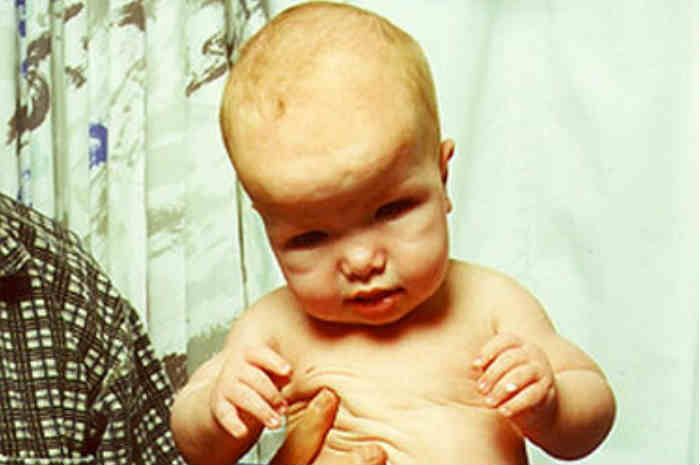
Manifestazione di sindrome di Sotos: macrocefalia
La Sindrome di Sotos è una malattia genetica rara caratterizzata da una crescita eccessiva durante il periodo infantile, un aspetto particolare del viso, macrocefalia (circonferenza cranica maggiore rispetto ai limiti per età), difficoltà di apprendimento o sviluppo ritardato delle capacità motorie e mentali.
La Sindrome di Sotos colpisce in egual misura maschi e femmine e indistintamente in qualsiasi gruppo etnico. L’esatta prevalenza della malattia non è nota, poiché la sindrome si presenta con una sintomatologia simile a quella di altre condizioni patologiche. Tuttavia è stimata intorno a 1-9/100.000 nati vivi.
Cause della sindrome di Sotos
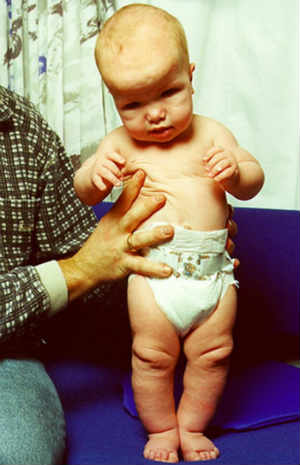
La Sindrome di Sotos è causata da mutazioni o delezioni (parziali o totali) del gene NSD1, localizzato sul cromosoma 5q35.
Il gene responsabile, in assenza di mutazioni, produce una proteina che regola l’attività di un gruppo di geni coinvolti nei processi di crescita e sviluppo del corpo umano; la sua funzione è quella di evitare l’iper-proliferazione cellulare.
Al contrario, quando alterato, il gene perde la sua funzione regolatrice, con conseguente crescita corporea eccessiva, soprattutto in altezza e dimensioni della testa.
La malattia viene trasmessa per lo più con modalità autosomica dominante: è cioè sufficiente che la mutazione riguardi una sola copia del gene responsabile.
Nel 10% dei casi la mutazione viene trasmessa proprio con questa modalità da un genitore affetto. Nel restante 90% dei casi la mutazione insorge in modo spontaneo (o de novo), cioè compare dal nulla nel corso dello sviluppo embrionale (dopo che la cellula uovo è stata fecondata dallo spermatozoo). In questo caso la mutazione è presente solo nella persona affetta e non ci sono altri casi familiari, se non raramente (meno del 2%).
Una persona affetta ha un rischio del 50% di trasmettere la malattia al proprio figlio ad ogni gravidanza, anche se la sintomatologia può variare sia all’interno della stessa famiglia sia in presenza dello stesso tipo di mutazione.
Se un individuo riceve invece dai genitori un gene normale e uno mutato, sarà portatore sano della malattia e non mostrerà la sintomatologia caratteristica. Il rischio per un individuo portatore sano di trasmettere la malattia alla prole è del 25% a ogni gravidanza.
Quali sono i sintomi della sindrome di Sotos
I sintomi della Sindrome di Sotos si manifestano sin dalla nascita e sono una crescita superiore alla norma (che inizia già nel periodo pre-natale) e una particolare configurazione cranio-facciale, che solo in una minima percentuale di casi non è presente.
Caratteristiche fisiche
Circa il 90% dei bambini affetti dalla Sindrome di Sotos ha un’altezza superiore alla media e a causa dell’eccessiva crescita scheletrica durante l’infanzia, in età adulta viene raggiunta un’altezza non inferiore a 1,93 m. Il peso è invece sovrapponibile a quello di individui sani della stessa altezza.
L’iper-accrescimento è più marcato nei primi 3-4 anni di vita per stabilizzarsi intorno a livelli più normali dopo i 5 anni di età. Anche la maturazione ossea nei primi anni di vita è anticipata, tanto che gli individui affetti dalla Sindrome di Sotos hanno solitamente un’età ossea di 2-4 anni superiore a quella dei loro coetanei sani.
Caratteristiche cranio-facciali
Per quanto riguarda le anomalie cranio-facciali, la testa appare molto grande, con valori superiori al 97° percentile e allungata in senso anteroposteriore (dolicocefalia).
Sono presenti, inoltre:
- palato ogivale (stretto e allungato verso l’alto)
- mandibola stretta e prominente
- mento appuntito
- fronte ampia e prominente
- impianto alto dei capelli a livello della fronte e capelli radi nella regione fronto-parietale
- fessure palpebrali rivolte verso il basso
- occhi distanziati tra loro in modo anomalo (ipertelorismo).
Le anomalie cranio-facciali sono generalmente molto più evidenti durante l’infanzia per attenuarsi poi con la crescita. Durante l’età adulta, infatti, gli unici segni ancora chiaramente riscontrabili sono la dolicocefalia, mento e fronte prominenti e attaccatura alta dei capelli.
Ritardo nello sviluppo intellettivo e motorio
L’acquisizione delle principali tappe dello sviluppo intellettivo (conseguenza di problemi a livello del sistema nervoso centrale) e motorio nelle persone con Sindrome di Sotos è ritardata.
Si riscontrano infatti ipotonia muscolare, lassità delle articolazioni, difficoltà nell’imparare a gattonare, a stare seduti, a muovere i primi passi, ma anche nella coordinazione del movimento, con conseguente goffaggine e difficoltà in attività come l’andare in bici o afferrare piccoli oggetti.
Queste difficoltà, ad eccezione della difficoltà di imparare a camminare (generalmente non camminano prima dei 15-17 mesi), caratteristica della prima infanzia, permangono tutta la vita.
Il ritardo intellettivo si manifesta fondamentalmente con un’acquisizione del linguaggio particolarmente lenta (potrebbero non iniziare a parlare fino ai 2-3 anni circa), come anche balbuzie, voce monotona e problemi nella riproduzione del suono.
Altri problemi associati alla sindrome di Sotos
Possono inoltre essere presenti iperattività, deficit di attenzione, aggressività, fobie, irritabilità e disturbi ossessivi-compulsivi, che rendono difficile lo sviluppo di relazioni interpersonali.
Ma anche anomalie cerebrali, scoliosi, anomalie genito-urinarie, malformazioni cardiache, crisi epilettiche, perdita di udito, problemi di vista, maggiore frequenza di infezioni delle vie respiratorie superiori e strabismo.
In epoca neonatale possono essere presenti anche ittero e difficoltà di alimentazione, mentre più avanti può esserci una prematura eruzione dei denti.
Nei bambini con Sindrome di Sotos è lievemente aumentato (2%) anche il rischio di tumori, per lo più neuroblastoma, ganglioma presacrale, teratoma sacro coccigeo, carcinoma polmonare a piccole cellule e leucemia linfoblastica acuta.
Diagnosi di Sindrome di Sotos
Il sospetto clinico si basa sulla raccolta della storia clinica e dell’evidenziazione dei sintomi caratteristici subito dopo la nascita, soprattutto a causa delle caratteristiche cranio-facciali e la crescita eccessiva.
La conferma diagnostica si basa sull’analisi del DNA per dimostrare, mediante sequenziamento del genoma, la presenza di mutazioni specifiche del gene NSD1.
Le microdelezioni del cromosoma 5q35 e le delezioni parziali del gene che, nelle popolazioni occidentali costituiscono circa il 10-15% dei casi, possono invece essere rilevate mediante i test di analisi di ibridazione fluorescente in situ (FISH) e di amplificazione legatura-dipendente multipla della sonda (MLPA).
Nei pazienti che invece non presentano mutazioni del gene NSD1 sono necessari ulteriori test genetici per verificare la presenza di mutazioni nei geni NFIX e APC2. La consulenza genetica è inoltre raccomandata non solo per gli individui affetti, ma anche per le loro famiglie.
Al momento della diagnosi di Sindrome di Sotos, un bambino dovrebbe essere sottoposto a visita cardiologica e nefrologica per identificare e trattare precocemente eventuali anomalie. E nelle prime fasi dello sviluppo dovrebbero essere indagate la presenza e il grado di ritardo dello sviluppo e psicomotorio e/o della disabilità intellettiva.
Inoltre ogni uno o due anni questi bambini dovrebbero essere sottoposti a visita ortopedica, oculistica e a valutazione del linguaggio. E, a seconda delle necessità, a ulteriori valutazioni cliniche, al fine di intervenire precocemente e con misure adeguate ad aiutare gli individui a raggiungere il massimo potenziale possibile per loro.
Diagnosi differenziale
La diagnosi differenziale viene posta con malattie caratterizzate da sintomatologia simile, come la Sindrome di Weaver, la Sindrome di Beckwith-Wiedemann, la Sindrome dell’X fragile, la Sindrome da delezione 22q e la Sindrome di Simpson-Golabi-Behmel.
Come si tratta la sindrome di Sotos
Il trattamento è basato sui sintomi e la presa in carico è multidisciplinare. Dal pediatra, all’endocrinologo, al genetista, al cardiologo, al neurologo e così via. Oltre a un supporto fisioterapico, psicoterapeutico e logopedico.
Sono importanti quindi periodici follow-up, specialmente nei primi anni di vita per tenere sotto controllo anche eventuali complicanze.
È importante per lo sviluppo globale del paziente un adeguato programma psicologico e educativo in associazione alla terapia del linguaggio e alla stimolazione motoria. Ulteriori interventi utili potrebbero essere la stimolazione infantile, la terapia occupazionale, la logopedia, l’attività fisica adattiva e l’educazione speciale.














Commento (0)
Devi fare il login per lasciare un commento. Non sei iscritto ?